This website is intended for UK healthcare professionals only. Not a UK healthcare professional? Click here.
Efficacy
Key efficacy information for NUSTENDI®/NILEMDO® is reported below. For full information on clinical trials, please refer to the relevant trial publications signposted under references and always refer to the SmPC before prescribing. For important safety information, please refer to the safety profile resources for NUSTENDI®/NILEMDO®.
In a post-hoc analysis, NUSTENDI® delivered significant LDL-C reductions on top of maximally-tolerated statin therapy vs. placebo and vs. ezetimibe1,*
Concomitant use of NUSTENDI® with simvastatin >40 mg daily is contraindicated; please refer to the SmPC for more information.2
Study 053 aimed to evaluate the efficacy and safety of NUSTENDI® vs. placebo, or ezetimibe or bempedoic acid alone in patients with hypercholesterolaemia and a high risk of cardiovascular disease receiving maximally tolerated statin therapy.1
38% LDL-C reduction vs. placebo
(placebo-corrected) from baseline at 12 weeks1,*
p<0.001 (post-hoc result)
13% LDL-C reduction vs. ezetimibe
from baseline at 12 weeks1,*
p<0.001 (post-hoc result)
*Study 053 was a Phase 3, randomised, double-blind, multicentre study in patients (N=382) with ASCVD, HeFH or multiple CVD risk factors, taking maximally-tolerated statin therapy (which could be no statin). Patients were randomised to NUSTENDI® (180/10 mg), bempedoic acid (180 mg), ezetimibe (10 mg) or placebo for 12 weeks. The post-hoc efficacy population included 301 patients (NUSTENDI® n=86, bempedoic acid n=88, ezetimibe n=86, placebo n=41) due to the exclusion of 81 patients from 3 study sites because of data integrity concerns.1

Summary1
- Study 053 was a Phase 3, randomised, double-blind, placebo-controlled trial that enrolled 382 adult patients at high risk of CVD due to ASCVD, HeFH or multiple CVD factors*
- After a screening period, 382 patients were randomly assigned 2:2:2:1 to oral, once-daily treatment with NUSTENDI® (bempedoic acid 180 mg + ezetimibe 10 mg, fixed-dose combination tablet), bempedoic acid 180 mg, ezetimibe 10 mg or placebo added to stable background statin therapy for 12 weeks
- The post-hoc efficacy population included 301 patients due to the exclusion of 81 patients from 3 study sites because of data integrity concerns
- One patient in the NUSTENDI® group did not receive any dose of study drug and was excluded from the safety analysis
Study design1

Key inclusion criteria1
While receiving stable maximally tolerated statin therapy, LDL-C levels of:
- ≥2.6 mmol/L in patients with ASCVD and/or HeFH
- ≥3.4 mmol/L in patients with multiple CVD risk factors
Key exclusion criteria1,3
- Patients were prohibited from using systemic corticosteroids, simvastatin at doses of 40 mg/day or greater, fibrates, niacin or derivatives, bile acid sequestrants, PCSK9 inhibitors, mipomersen, lomatipide, cholesteryl ester transfer protein inhibitors, or red yeast rice- containing products or undergoing apheresis during the study or within specified intervals before screening
- Uncontrolled hypertension: systolic blood pressure ≥160 mmHg and/or diastolic blood pressure ≥100 mmHg
- Liver disease or dysfunction, including alanine aminotransferase (ALT), aspartate aminotransferase (AST) ≥2× ULN, and/or total bilirubin (TB) ≥2× ULN at Week -2
- eGFR <30 mL/min/1.73 m2
- Total fasting triglyceride ≥5.6 mmol/L
- BMI ≥40 kg/m2
Endpoints1
- Primary endpoint: % change in LDL-C levels from baseline to Week 12
- Co-primary endpoints: Comparisons between the NUSTENDI® and 3 other treatment arms (placebo, ezetimibe and bempedoic acid monotherapy)
- Key secondary endpoints: % change in non-LDL-C levels and total cholesterol from baseline to Week 12
Results1
Safety1
- Safety was assessed by continuous monitoring of TEAEs as well as clinical laboratory values, vital sign measurements, weight changes, physical examination findings and electrocardiogram readings
- Safety and tolerability findings were consistent with expectations based on previous bempedoic acid clinical trials (see NILEMDO® trials below)
- TEAEs were more frequently observed in the NUSTENDI® and bempedoic acid groups than in the ezetimibe or placebo groups
- The most common TEAEs in the NUSTENDI® group were blood uric acid increase, constipation, fatigue, muscle spasms and oral discomfort, each reported by 2.4% (n=2) patients
- Rates of individual AEs were low (affecting <7% of patients per treatment group), and were determined to be mild or moderate in intensity
- The rate of AE-related discontinuations was similar in all active treatment groups but higher vs. placebo (NUSTENDI® 8.2% [n=7]; bempedoic acid 10.2% [n=9]; ezetimibe 11.6% [n=10]; placebo 4.9% [n=2])
- AEs leading to discontinuation in active treatment groups included myalgia; bempedoic acid 3.4% [n=3]; ezetimibe 1.2% [n=1]); oral discomfort (NUSTENDI® 2.4% [n=2]); fatigue (NUSTENDI® 1.2% [n=1]; bempedoic acid 1.1% [n=1])
*Multiple CVD risk factors was defined as diabetes plus one other risk factor or 3 CVD risk factors from the following list: age (men ≥45 years, women ≥55 years); family history of CHD; smoking; hypertension; low HDL-C; or coronary calcium score above the 95th percentile for the patient’s age, sex, and race/ethnicity.1
NILEMDO® has demonstrated efficacy in reducing elevated LDL-C across hyperlipidaemia studies4–7
Concomitant use of NILEMDO® with simvastatin >40 mg daily is contraindicated; please refer to the SmPC for more information.8
NILEMDO® delivered a significant
17–28% LDL-C reduction vs. placebo
(placebo-corrected) from baseline at 12 weeks, depending on risk factors and concomitant medicine4–7,*
For details and results of individual studies, please browse the accordions below.
*Placebo-corrected LDL-C reductions in pivotal NILEMDO® studies: CLEAR Harmony, 18.1%, n=1,488; CLEAR Wisdom, 17.4%, n=522; CLEAR Serenity, 21.4%, n=234; CLEAR Tranquility, 28.5%, n=181. All p<0.001 for NILEMDO® vs. placebo. CLEAR Harmony and CLEAR Wisdom included patients with ASCVD, HeFH or both, taking maximally-tolerated statins (which could be no statin) +/- LLT. CLEAR Serenity included primary and secondary prevention patients with statin intolerance taking very-low dose statin, non-statin LLT or no LLT. CLEAR Tranquility included primary and secondary prevention patients with statin intolerance taking ezetimibe with low dose, very-low dose or no statin +/- other non-statin LLT.4–7
Please expand the sections below for more details on each specific study.
Summary4
- The CLEAR Harmony trial was a Phase 3, randomised, double-blind, placebo-controlled trial that enrolled patients with ASCVD, HeFH, or both
- After a 2-week screening period, 2,230 patients were randomly assigned 2:1* to oral once-daily NILEMDO® 180 mg or placebo once daily for 52 weeks
- One patient underwent randomisation in error and was excluded from the safety population
Study composition4
~93% of patients were treated with moderate- or high-intensity statins
~8% of patients were using ezetimibe
Study design4

Key inclusion criteria4
- Adults with ASCVD, HeFH or both
- On maximally-tolerated statin therapy† with or without other LLTs‡ for at least 4 weeks before screening
- Fasting LDL-C levels of ≥1.8 mmol/L during screening period
Key exclusion criteria4,9
- Use of gemfibrozil or simvastatin at doses greater than 40 mg per day (although high-intensity atorvastatin and rosuvastatin regimens were permitted)
- Use of any inhibitor of proprotein convertase subtilisin-kexin type 9 (PCSK9) was prohibited starting 4 weeks before trial entry but was permitted after trial Week 24 if the LDL-C level was greater than 170 mg/dL (4.4 mmol/L) and had increased by at least 25% from baseline
- Uncontrolled hypertension: systolic blood pressure ≥160 mmHg and/or diastolic blood pressure ≥100 mmHg
- Liver disease or dysfunction, including alanine aminotransferase (ALT), aspartate aminotransferase (AST) ≥2× ULN, and/or total bilirubin (TB) ≥1.2× ULN at Week -2
- eGFR <30 mL/min/1.73 m2 (or renally impaired patients receiving an average daily dose of simvastatin 40 mg with eGFR below <45 mL/min/1.73 m2)
- Total fasting triglyceride ≥500 mg/dL (5.6 mmol/L)
- BMI >50kg/m2
Endpoints4
- Primary endpoint (safety): Overall safety, assessed according to the incidence of AEs and changes in safety laboratory variables from receipt of the first dose through 30 days after the last dose
- Principal secondary endpoint (efficacy): % change in LDL-C levels from baseline to Week 12
- Additional key secondary endpoints: % changes in non-HDL-C levels, total cholesterol from baseline to Week 12
Results4
Safety4
- The incidence of adverse events and serious adverse events was similar between the NILEMDO® and placebo groups: Adverse events (bempedoic acid 78.5% [n=1167]; placebo 78.7% [n=584]); Serious adverse events (bempedoic acid 14 .5% [n=216]; placebo 14.0 % [n=104])
- The incidence of adverse events leading to discontinuation of the regimen was higher in the bempedoic acid group than in the placebo group (10.9 % [n=162] vs. 7.1% [n=53]), as was the incidence of gout (1.2 % [n=18] vs. 0.3% [n=2])
LDL-C4
- NILEMDO® delivered a significant 18.1% (95% CI: 20.0% to 16.1%) LS mean LDL-C reduction (placebo-corrected) vs. placebo at Week 12 (p<0 .001) on top of maximally tolerated statins† alone or in combination with other LLTs
- The effects of bempedoic acid on LDL-C were still apparent through Week 52 with minimal attenuation of effect in the on-treatment analyses
*Randomisation stratified by baseline statin intensity (low-, moderate-, or high-intensity) and HeFH (present or absent).4
†Defined as the highest intensity statin regimen that the patient was able to maintain, as determined by the investigator.4
‡Other LLTs could include ezetimibe or fibrates. The use of PCSK9 inhibitors was prohibited starting 4 weeks before trial entry but was permitted after trial Week 24 if the LDL-C level was greater than 4.4 mmol/L and had increased by at least 25% from baseline.4
Summary5
- The CLEAR Wisdom trial was a Phase 3, randomised, double-blind, placebo-controlled trial that enrolled patients at high CV risk due to ASCVD and/or HeFH*
- The CLEAR Wisdom trial comprised a 1-week screening period and a 4-week placebo run-in phase, 779 patients were randomly assigned 2:1† to oral once-daily NILEMDO® 180 mg or placebo for 52 weeks
Study composition5
~89% of patients were treated with statins as background therapy
~85% were treated with moderate-or high-intensity statins
~8% of patients were using ezetimibe
Study design5

Key inclusion criteria5
- While receiving maximally-tolerated LLT:‡,§
- Fasting levels of:
- ≥2.6 mmol/L at the first screening
- ≥1.8 mmol/L 1 week before randomisation
Key exclusion criteria5,10
- Maximally tolerated LLT was determined by the investigator and included a maximally tolerated statin dose alone or in combination with other LLTs, excluding simvastatin at an average daily dose of 40 mg or greater, mipomersen (unlicensed in the UK), lomitapide, lipoprotein apheresis, or gemfibrozil
- Uncontrolled hypertension: systolic blood pressure ≥160 mmHg and/or diastolic blood pressure ≥100 mmHg
- Liver disease or dysfunction, including alanine aminotransferase (ALT), aspartate aminotransferase (AST) ≥2x ULN and/or total bilirubin (TB) ≥2x ULN at Week -5
- eGFR <30 mL/min/1.73 m2
- Total fasting triglyceride ≥5.6 mmol/L (500 mg/dL)
- BMI ≥50 kg/m2
Endpoints5
- Primary endpoint: % change in LDL-C levels from baseline to Week 12
- Secondary endpoints: % change in non-HDL-C levels and total cholesterol from baseline to Week 12
Results5
LDL-C5
- NILEMDO® delivered a significant 17.4% (95% CI: 21.0% to 13.9%) LS mean LDL-C reduction (placebo-corrected) from baseline to Week 12 (p<0.001) on top of maximally tolerated LLTs
Safety5
- Safety was assessed by continuous monitoring of TEAEs as well as clinical laboratory values, vital sign measurements, weight changes, physical examination findings and electrocardiogram readings
- No difference in overall TEAEs between NILEMDO® (70.1% [n=366]) and placebo (70.8% [n=182]) at Week 52
- The majority of adverse events across both treatment groups (78.5% [n=430]) were mild or moderate in intensity, and most (77.6% [n=425]) were classified as not related or unlikely related to study drug treatment
- TEAEs leading to discontinuation occurred in 10.9% (n=57) of paients taking NILEMDO® vs. 8.6% (n=22) taking placebo
*ASCVD included CHD (acute MI, silent MI, unstable angina, coronary revascularisation or clinically significant CHD diagnosed by invasive or non-invasive testing) or CHD risk equivalents (cerebrovascular atherosclerotic disease or symptomatic peripheral arterial disease).5
†Randomisation was stratified by presence of HeFH and baseline statin intensity (low-, moderate-, or high-intensity).5
‡Determined by the investigator and included a maximally-tolerated statin dose alone or in combination with other approved LLTs, excluding simvastatin at an average daily dose of ≥40 mg, mipomersen (unlicensed in UK), lomitapide, lipoprotein apheresis, or gemfibrozil. Background LLT dose could be adjusted or additional LLT added by the investigator from Week 24 if the LDL-C was >4.4 mmol/L and elevated ≥25% from baseline.5
§Defined as the highest intensity statin regimen that the patient was able to maintain, as determined by the investigator.5
Summary6
- The CLEAR Serenity trial was a Phase 3, randomised, double-blind, parallel-group, placebo-controlled trial that enrolled 345 patients with hypercholesterolaemia and a history of intolerance to at least two statins (1 at the lowest dose)
- After a 5-week screening period (including a 4-week, single-blind, placebo run-in period), 345 patients were randomly assigned 2:1* to oral once-daily NILEMDO® 180 mg or placebo for 24 weeks
- Patients were permitted the following background LLT if initiated ≥4 weeks prior to screening: ezetimibe, bile acid sequestrants, fibrates (except gemfibrozil in patients receiving a very-low-dose statin), PCSK9 inhibitors (if ≥3 doses were received prior to screening), niacin, or very-low-dose statin therapy (average daily dose of rosuvastatin <5 mg, atorvastatin <10 mg, simvastatin <10 mg, lovastatin <20 mg (unlicensed in UK), pravastatin <40 mg, fluvastatin <40 mg, or pitavastatin <2 mg (unlicensed in UK), either alone or in combination
Study composition6
8.4% of patients were treated with very low dose statin
1/3 of patients (n=116) were treated with non-statin LLTs (including ezetimibe)
Study design6

Key inclusion criteria6
While receiving stable background LLT:†
- Fasting LDL-C levels of ≥2.6 mmol/L in patients with HeFH and/or a secondary prevention indication‡
- Fasting LDL-C levels of ≥3.4 mmol/L in primary prevention patients
- A history of statin intolerance (to at least 2 statins including 1 at a low dose)§,¶
Key exclusion criteria6,11
- Uncontrolled hypertension: systolic blood pressure ≥160 mmHg and/or diastolic blood pressure ≥100 mmHg
- eGFR <30 mL/min/1.73 m2
- Liver disease or dysfunction, including alanine aminotransferase (ALT) ≥2× ULN, aspartate aminotransferase (AST) ≥2× ULN, and/or total bilirubin (TB) ≥1.2× ULN at Week -5
- Total fasting triglyceride ≥5.6 mmol/L (500 mg/dL)
- BMI ≥50 kg/m2
Endpoints6
- Primary endpoint: % change in LDL-C levels from baseline to Week 12
- Secondary endpoints: % change in LDL-C levels from baseline to Week 24 and % change in non-LDL-C levels and total cholesterol from baseline to Week 12
Results6
LDL-C6
- NILEMDO® delivered a significant 21.4% (95% CI: 25.1% to 17.7%) LDL-C LS mean reduction vs. placebo (placebo-corrected) from baseline at Week 12 (p<0.001) in statin-intolerant patients
Safety6
- Safety was assessed by continuous monitoring of TEAEs, clinical safety laboratory findings, vital sign measurements, physical examinations, electrocardiograph readings, and cardiovascular event rates
- Overall rates of TEAEs were 64.1% (n=150) in the NILEMDO® group vs. 56.8% (n=63) in the placebo group at Week 24
- The majority of AEs in both the NILEMDO® and placebo groups were mild to moderate in intensity
- TEAEs leading to discontinuation occurred in 18.4% (n=43) in the NILEMDO® group vs. 11.7% (n=13) in the placebo group at Week 24
*Randomisation was stratified by treatment indication (primary prevention vs. secondary prevention and/or HeFH).6
†Patients were permitted the following background LLT if initiated ≥4 weeks prior to screening: ezetimibe, bile acid sequestrants, fibrates (except gemfibrozil in patients receiving a very-low-dose statin), PCSK9 inhibitors (if ≥3 doses were received prior to screening), niacin, or very-low-dose statin therapy (average daily dose of rosuvastatin <5 mg, atorvastatin <10 mg, simvastatin <10 mg, lovastatin <20 mg (unlicensed in UK), pravastatin <40 mg, fluvastatin <40 mg, or pitavastatin <2 mg (unlicensed in UK), either alone or in combination.6
‡Including coronary artery disease, symptomatic peripheral arterial disease, and/or cerebrovascular atherosclerotic disease.6
§Average daily dose of rosuvastatin 5 mg, atorvastatin 10 mg, simvastatin 10 mg, lovastatin 20 mg (unlicensed in UK), pravastatin 40 mg, fluvastatin 40 mg, or pitavastatin 2 mg (unlicensed in UK).6
¶Due to a prior adverse event that started or increased during statin therapy and resolved or improved when statin therapy was discontinued.6
Summary7
- The CLEAR Tranquility trial was a Phase 3, randomised, double-blind, placebo-controlled trial that enrolled 269 patients with an LDL-C ≥2.6 mmol/L while on no more than low-dose statin therapy (which could include no statin) and with a history of statin intolerance (not tolerating at least one statin) requiring additional LDL-C lowering
- During a 4-week run-in phase, patients received open-label ezetimibe 10 mg once daily and single-blind placebo to confirm tolerance to ezetimibe and compliance with protocol-directed therapy
- After the run-in period, individuals were randomised 2:1 to treatment with oral once-daily NILEMDO® 180 mg or placebo added to once-daily ezetimibe 10 mg/day for 12 weeks
Study composition7
~31% of patients (32.6% NILEMDO; 27.6% placebo) were receiving stable background lipid-modifying therapy (inclusive of a low-dose or very low-dose statin and/or permitted non-statin agents)
100% of patients were treated with study-provided open-label ezetimibe 10 mg once daily, maintained throughout the study
Study design7

Key inclusion criteria7
While receiving low-dose statin therapy,† which could also include no statin:
- Fasting LDL-C levels of ≥2.6 mmol/L at screening
- A history of statin intolerance (to at least 1 statin)
Key exclusion criteria7,12
- Liver disease or dysfunction, including: alanine aminotransferase (ALT), aspartate aminotransferase (AST) ≥2× ULN, and/or total bilirubin (TB) ≥2× ULN at Week -5
- Uncontrolled hypertension: systolic blood pressure ≥160 mmHg and/or diastolic blood pressure ≥100 mmHg
- eGFR <30 mL/min/1.73 m2
- Total fasting triglyceride ≥5.6 mmol/L (500 mg/dL)
- BMI ≥50 kg/m2
Endpoints7
- Primary endpoint: % change in LDL-C levels from baseline to Week 12
- Secondary endpoints: % change in non-HDL-C levels and total cholesterol from baseline to Week 12
Results7
LDL-C7
- NILEMDO® delivered a significant 28.5% (95% CI: 34.4% to 22.5%) LS mean LDL-C reduction vs. placebo (placebo-corrected) from baseline to Week 12 (p<0.001) in statin-intolerant patients taking ezetimibe +/- other LLT
Safety7
- Safety was assessed by continuous monitoring of TEAEs, clinical safety laboratory findings, vital sign measurements, physical examinations, electrocardiograph readings, and weight measurements and were judged to be not related or unlikely related to investigational study drug
- TEAEs were reported by 48.6% (n=88) and 44.8% (n=39) of patients in the NILEMDO® and placebo groups, respectively
- The majority of TEAEs in the NILEMDO® group and the placebo group were mild to moderate in intensity and were judged to be not related or unlikely related to investigational study drug
- Serious TEAEs occurred in 2.8% (n=5) and 3.4% (n=3) of patients in the bempedoic acid and placebo treatment groups, respectively
- TEAEs leading to discontinuation occurred in 6.1% (n=11) in the NILEMDO® group vs. 5.7% (n=5) in the placebo group
*Patients with ≤80% adherence and/or who experienced a study drug-related adverse event during the placebo and ezetimibe run-in period did not proceed to randomisation.7
†Low-dose statin therapy was defined as an average daily dose of rosuvastatin 5 mg, atorvastatin 10 mg, simvastatin 10 mg, lovastatin 20 mg (unlicensed in UK), pravastatin 40 mg, fluvastatin 40 mg, or pitavastatin 2 mg (unlicensed in UK).7
Bempedoic acid significantly reduced major CV events vs. placebo in the CLEAR Outcomes trial13
Concomitant use with simvastatin >40 mg daily is contraindicated; please refer to the relevant SmPC for more information2,8
CLEAR Outcomes did not assess NUSTENDI® (bempedoic acid + ezetimibe).13 Evidence for the use of NUSTENDI® in patients at high risk of ASCVD is only available for the lipid-lowering effect (see section 5.1 of the NUSTENDI® SmPC).2
CLEAR Outcomes is a randomised, double-blind, placebo-controlled Phase 3 trial to assess the effects of bempedoic acid on the occurrence of major CV events in patients with, or at high risk for, CVD who are unable or unwilling to take guideline-recommended doses of statins.13,*
13% Reduction in Relative Risk
1.6% ARR p=0.004 in first-event MACE-413
MACE-4: four-component composite of major adverse CV events defined as death from CV causes, non-fatal MI, non-fatal stroke or coronary revascularisation.13
*Patients were permitted the following background LLT without unacceptable adverse effects if initiated ≥4 weeks prior to screening: ezetimibe, niacin, bile acid resins, fibrates and/or PCSK9 inhibitors, or very-low-dose statin therapy (average daily dose of rosuvastatin <5 mg, atorvastatin <10 mg, simvastatin <10 mg, lovastatin <20 mg (unlicensed in UK), pravastatin <40 mg, fluvastatin <40 mg, or pitavastatin <2 mg (unlicensed in UK), either alone or in combination).13,14
CLEAR Outcomes key secondary
endpoint
CLEAR Outcomes LDL-C
reduction
After 6 months, over the remaining duration of the trial, 15.6% of patients in the placebo group received additional LLT, compared with 9.4% in the bempedoic acid group.13
At 6 months after randomisation, investigators were notified if LDL-C level was 25% or higher than baseline. These patients received dietary and lipid-regulating medication adherence counselling. If repeat testing confirmed that the LDL-C level exceeded the threshold, the investigator could adjust the lipid-lowering regimen according to standard of care and local guidelines. Investigators remained blinded to trial group and laboratory values.13

Summary13
- A randomised, double-blind, placebo-controlled Phase 3 trial to assess the effects of bempedoic acid on the occurrence of major CV events in patients with, or at high risk for, CVD who are unable or unwilling to take guideline-recommended doses of statins
Study design13
*All patients provided written informed consent. Eligible patients had to report being unable or unwilling to receive statins owing to an adverse effect that had started or increased during statin therapy and resolved or improved after statin therapy was discontinued (“statin-intolerant” patients). The patients were required to provide written confirmation that they were statin intolerant and aware of the benefits of statins in reducing the risk of CV events, including death, as well as acknowledge that many patients who are unable to receive an administered statin can receive a different statin or dose; the site investigators were also required to confirm and acknowledge these statements with respect to the patients.13
†Patients were permitted the following background LLT without unacceptable adverse effects if initiated ≥4 weeks prior to screening: ezetimibe, niacin, bile acid resins, fibrates and/or PCSK9 inhibitors, or very-low-dose statin therapy (average daily dose of rosuvastatin <5 mg, atorvastatin <10 mg, simvastatin <10 mg, lovastatin <20 mg (unlicensed in UK), pravastatin <40 mg, fluvastatin <40 mg, or pitavastatin <2 mg (unlicensed in UK), either alone or in combination.13
‡Including CV death, non-fatal MI, non-fatal stroke or coronary revascularisation.13
§Including CV death, non-fatal MI or non-fatal stroke.13
Primary efficacy endpoint13
- The primary endpoint was a four-component composite of major adverse CV events (MACE-4), defined as death from CV causes, non-fatal MI, non-fatal stroke or coronary revascularisation, as assessed in a time-to-first event analysis
Key secondary time-to-event endpoints13
- A three-component composite of major adverse CV events (MACE-3) defined as death from CV cause, non-fatal MI or non-fatal stroke
- Fatal or non-fatal MI
- Coronary revascularisation
- Fatal or non-fatal stroke
- Death from CV causes
- Death from any cause
Key inclusion criteria13
- A previous CV event (secondary-prevention patients) or clinical features that placed patients at high risk for a CV event (primary-prevention patients)
- Unable or unwilling to tolerate statins due to an adverse event that started or increased during statin therapy and resolved or improved when statin therapy was discontinued (“statin intolerance”)
- Written confirmation by both patient and investigator that the patient is statin intolerant as defined above, aware of the benefit of statin use to reduce the risk of MACE including death, and also aware that many other patients who are unable to tolerate a statin are able to tolerate a different statin or dose
Key exclusion criteria13,14
- Forms of CVD that include recent (90 days prior to or during screening) acute CVD events including transient ischemic attack (TIA), MI, coronary revascularisation, peripheral arterial revascularisation, ischaemic stroke, carotid endarterectomy, carotid stenting; recent (90 days of screening) unstable or symptomatic cardiac arrhythmia; NYHA Functional Classification Class IV heart failure, uncontrolled hypertension: systolic blood pressure ≥180 mmHg and/or diastolic blood pressure ≥110 mmHg; planned coronary revascularisation
- Liver disease or dysfunction, including alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) ≥2x ULN at Week 5
- eGFR <30 mL/min/1.73 m2 at Week 5
- Total fasting triglyceride >500 mg/dL (5.6 mmol/L) at Week -5
Baseline change in secondary lipid and biomarker efficacy endpoints13
- Percent change in LDL-C and hsCRP
- Absolute change in HbA1c in patients with diabetes and HbA1c ≥7% at baseline
- Development of new diagnosis of diabetes in patients with prediabetes at baseline
Results13
Safety (Safety population N=13,965)13
- The safety population included all patients who underwent randomisation and received at least one dose of bempedoic acid or placebo; patients who received any dose of double-blind bempedoic acid were placed in the bempedoic acid group in the safety analyses
- No difference in the overall incidence of AEs between bempedoic acid 86.3% (n=6,040/7,001) and placebo 85% (n=5,919/6,964)
- AEs leading to discontinuation occurred in 10.8% (n=759/7,001) of patients receiving bempedoic acid vs. 10.4% (n=722/6,964) receiving placebo
- Incidences of prespecified AEs of special interest were similar between bempedoic acid and placebo, except for myalgia (5.6% [n=393/7,001] vs. 6.8% [n=471/6,964], respectively), elevations in hepatic enzyme levels (4.5% [n=317/7,001] vs. 3.0% [n=209/6,964], respectively) and renal impairment (11.5% [n=802/7,001] vs. 8.6% [n=599/6,964], respectively)
- The incidence of hyperuricemia was higher in the bempedoic acid group than in the placebo group (10.9% [n=763] vs. 5.6% [n=393], respectively), as were the incidences of gout (3.1% [n=215/7,001] vs. 2.1% [n=143/6,964], respectively) and cholelithiasis (2.2% [n=152/7,001] vs. 1.2% [n=81/6,964], respectively)
Table

Adapted from Nissen SE, et al. 2023.13
The effects of bempedoic acid in patients at high-risk of a first CV event were assessed in a prespecified sub-analysis of the CLEAR Outcomes trial15
Concomitant use with simvastatin >40 mg daily is contraindicated; please refer to the relevant SmPC for more information2,8
The data below was generated in the primary prevention prespecified sub-analysis of the CLEAR Outcomes trial.15
Primary prevention sub-analysis safety data.
CLEAR Outcomes assessed bempedoic acid only, the combination product bempedoic acid + ezetimibe (NUSTENDI®) was not assessed in this trial.13
Subgroup analyses should be considered as hypothesis generating only.
Prespecified population15
- Primary prevention patients aged 18 to 85 years with an LDL-C level 100 mg/dL (2.59 mmol/L) or greater and with clinical features placing them at high risk for a first CV event. (Coronary artery calcium score greater than 400 Agatson units, or presence of either T1 or T2DM in women older than 65 years or men older than 60 years or Reynolds Risk score >30% or SCORE Risk score >7.5% over 10 years)
- Within the full CLEAR Outcomes analysis population, 4206 patients (30%) met primary prevention criteria. Patients were randomised 1:1 to receive bempedoic acid (n=2100) or placebo (n=2106)

*Race and ethnicity were self-reported. Participants could select more than one category.15
†At baseline, medical history of type 1 or 2 diabetes, prior glucose-lowering medication, haemoglobin A1c level 6.5% (48 mmol/mol) or greater, or 2 or more measurements of fasting glucose 7.0 mmol/L or greater.16
‡Patients could be enrolled if they were taking doses of a statin less than the lowest approved dose16
In CLEAR Outcomes, bempedoic acid reduced first MACE-4 event vs. placebo in the primary prevention prespecified population15
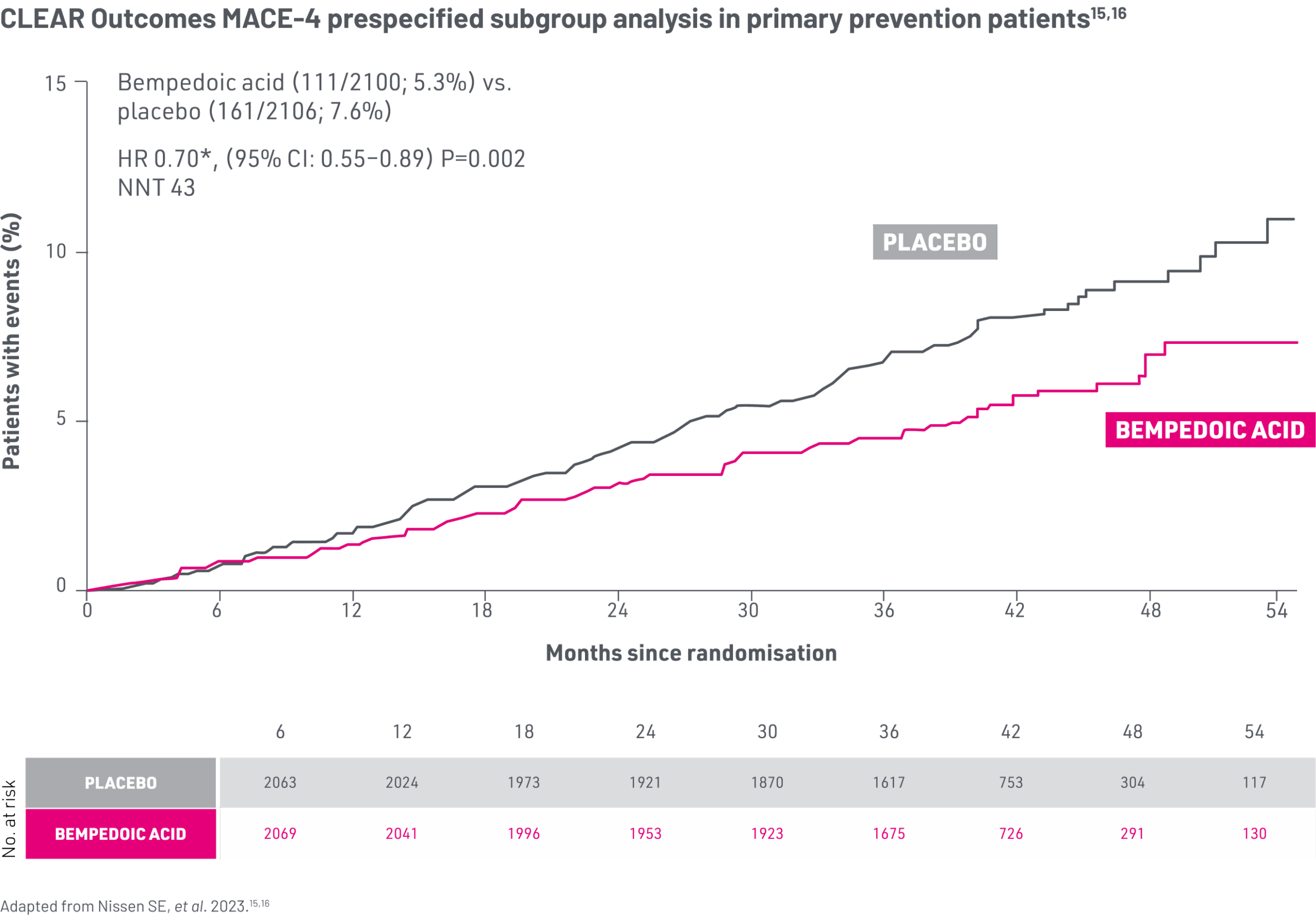
*Hazard ratios adjusted for baseline characteristics including geographic region, age, gender, race, ethnicity, baseline LDL-C, body mass index, high-sensitivity C reactive protein, estimated glomerular filtration rate, use of any lipid-modifying therapy at baseline and diabetes status (diabetes, prediabetes, normoglycaemia).15
30% Reduction in Relative Risk15
2.3% ARR p=0.002 in first-event MACE-4 (primary prevention subgroup)
MACE-4: four-component composite of major adverse CV events defined as death from CV causes, non-fatal MI, non-fatal stroke or coronary revascularisation.15

After 6 months, over the remaining duration of the study in the primary prevention subgroup, 12.4% of patients in the placebo group received additional LLT, compared with 6.7% in the bempedoic acid group.15
The effects of bempedoic acid in patients with diabetes were assessed in a prespecified sub-analysis of the CLEAR Outcomes trial17
Concomitant use with simvastatin >40 mg daily is contraindicated; please refer to the relevant SmPC for more information2,8
CLEAR Outcomes assessed bempedoic acid only, the combination product bempedoic acid + ezetimibe (NUSTENDI®) was not assessed in this trial.13
The data below was generated in the diabetes* prespecified sub-analysis of the CLEAR Outcomes trial.16
CLEAR Outcomes diabetes sub-analysis safety data.
Subgroup analyses should be considered as hypothesis generating only.
Prespecified population17
- At baseline, 45.6% (n=6373) patients were diabetic, 41.5% (n=5796) patients were prediabetic, and 12.9% (n=1801) patients were normoglycaemic*
Primary Efficacy endpoint17
- The primary endpoint was a four-component composite of major adverse CV events (MACE-4), defined as death from CV causes, non-fatal MI, non-fatal stroke or coronary revascularisation, as assessed in a time-to-first event analysis
Clinical endpoints17
- New onset diabetes in baseline prediabetic and normoglycaemic patients
- Mean change in HbA1c at 12 months in patients without diabetes (prespecified)
- Mean change in fasting glucose at 6 months in patients without diabetes (prespecified)
- Changes in HbA1c and fasting glucose were also examined at the end of the study (post-hoc analysis)
*Normoglycaemia: Did not meet the criteria for either prediabetes or diabetes. Pre-diabetes: HbA1c 5.7–6.4% (39–48 mmol/mol), or ≥1 fasting serum glucose concentration of at least 5.6 mmol/L, but with no more than one value of ≥7.0 mmol/L. Diabetes: Medical history of DM; or use of glucose lowering medication; or HbA1c >6.5% (48 mmol/mol); or two or more fasting serum glucose concentration ≥7.0 mmol/L.17
Patients with diabetes had a 25% higher risk of MACE-4 compared to individuals who were normoglycaemic in the CLEAR Outcomes trial17

*Normoglycaemia: Did not meet the criteria for either prediabetes or diabetes. Pre-diabetes: HbA1c 5.7–6.4% (39–48 mmol/mol), or ≥1 fasting serum glucose concentration of at least 5.6 mmol/L, but with no more than one value of ≥7.0 mmol/L. Diabetes: Medical history of DM; or use of glucose lowering medication; or HbA1c >6.5% (48 mmol/mol); or two or more fasting serum glucose concentration ≥7.0 mmol/L.17
Bempedoic acid demonstrated a reduction in CV risk for patients with diabetes in CLEAR Outcomes at Month 2417

*Over a median 3.4 years follow-up, in the placebo group, a graded relationship was observed for the incidence of the primary MACE-4 endpoint, which occurred in 11.9% of patients with normoglycaemia, 12.6% in patients with prediabetes and 14.2% in patients with diabetes (unadjusted HR 1.25, 95% CI: 1.01–1.55; p=0.042) for diabetes vs. normoglycaemia.17
Bempedoic acid provided similar relative but greater absolute benefits on MACE-4 in patients with diabetes compared to the overall patient CLEAR Outcomes population17
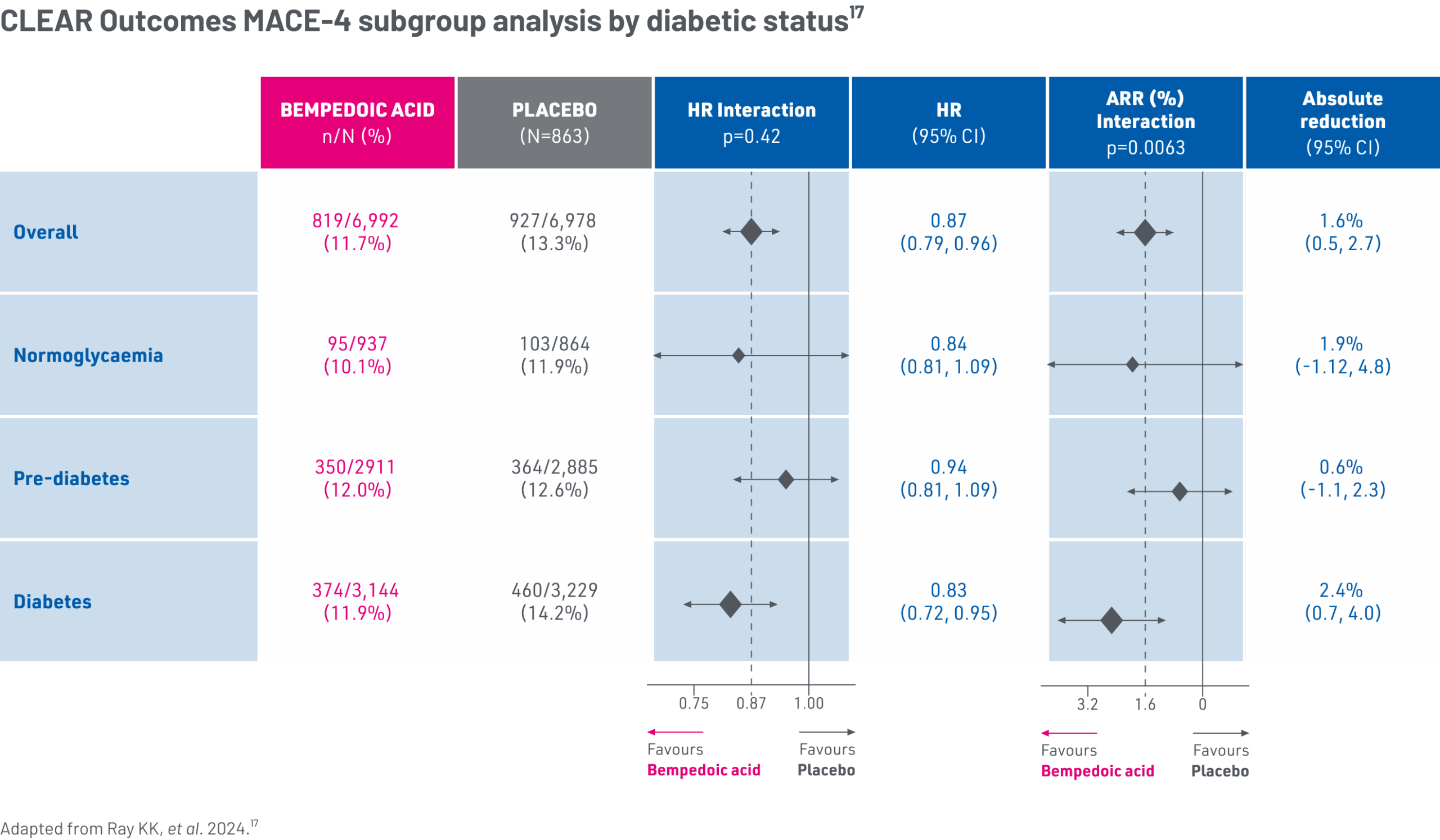
Due to higher baseline CV risk, the absolute risk reduction of bempedoic acid was greatest in patients with diabetes, compared to other glycaemic strata.17,*
In CLEAR Outcomes, bempedoic acid was not associated with new-onset diabetes vs. placebo17
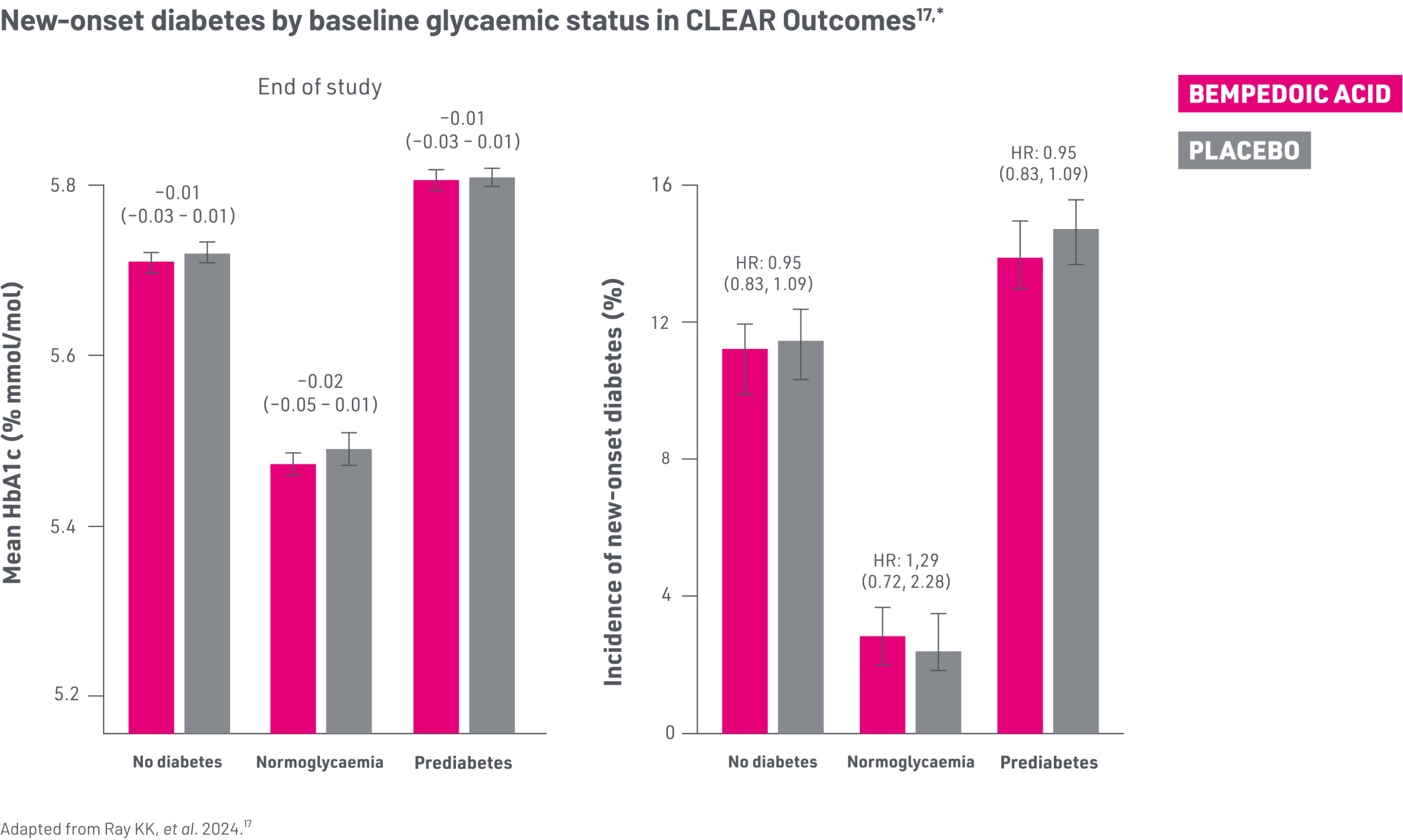
*Normoglycaemia: Did not meet the criteria for either prediabetes or diabetes. Pre-diabetes: HbA1c 5.7–6.4% (39–48 mmol/mol), or ≥1 fasting serum glucose concentration of at least 5.6 mmol/L, but with no more than one value of ≥7.0 mmol/L. Diabetes: Medical history of DM; or use of glucose lowering medication; or HbA1c >6.5% (48 mmol/mol); or two or more fasting serum glucose concentration ≥7.0 mmol/L.17
HbA1c concentrations at the end of the CLEAR Outcomes study were similar between randomised groups in patients who had prediabetes and normoglycaemia and there was no increase in the incidence of new-onset diabetes in patients treated with bempedoic acid vs. placebo after a median follow-up of 40.6 months.17
References
- Ballantyne CM, et al. Eur J Prev Cardiol. 2020;27:593–603.
- NUSTENDI® Summary of Product Characteristics.
- Ballantyne CM, et al. Eur J Prev Cardiol. 2020;27:593–603. Supplemental Protocol.
- Ray KK, et al. N Engl J Med. 2019;380:1022–1032.
- Goldberg AC, et al. JAMA. 2019;322:1780–1788.
- Laufs U, et al. J Am Heart Assoc. 2019;8:e011662.
- Ballantyne CM, et al. Atherosclerosis. 2018;277:195–203.
- NILEMDO® Summary of Product Characteristics.
- Ray KK, et al. N Engl J Med. 2019;380:1022–1032. Supplementary Appendix.
- Goldberg AC, et al. JAMA. 2019;322:1780–1788. Supplement 1. Study Protocol.
- Laufs U, et al. J Am Heart Assoc. 2019;8:e011662. Data S1. Supplemental Methods.
- Ballantyne CM, et al. Atherosclerosis. 2018;277:195–203. Supplementary Data.
- Nissen SE, et al. N Engl J Med. 2023;388(15):1353–1364.
- Nissen SE, et al. N Engl J Med. 2023;388(15):1353–1364. Supplementary Appendix.
- Nissen SE, et al. JAMA. 2023;330(2):131–140.
- Nissen SE, et al. JAMA. 2023;330(2):131–140. Supplementary online content.
- Ray KK, et al. Lancet Diabetes Endocrinol. 2024;12:19–28.
Abbreviations
AE, adverse event; ALT, alanine aminotransferase; ARR, absolute risk reduction; AST, aspartate aminotransferase; ASCVD, atherosclerotic cardiovascular disease; BMI, body mass index; CHD, coronary heart disease; CI, confidence interval; CV, cardiovascular; CVD, cardiovascular disease; DM, diabetes mellitus; eGFR, estimated glomerular filtration rate; HbA1c, haemoglobin A1c; HDL-C, high-density lipoprotein cholesterol; HeFH, heterozygous familial hypercholesterolaemia; HR, hazard ratio; hsCRP, high-sensitivity C-reactive protein; IQR, Interquartile Range; LDL-C, low-density lipoprotein cholesterol; LLT, lipid-lowering therapy; LS, least-squares; MACE, major adverse cardiac event; MI, myocardial infarction; NNT, number needed to treat; NYHA, The New York Heart Association; O.D., once a day; PCSK9, proprotein convertase subtilisin/kexin type 9; SD, standard deviation; SmPC, Summary of Product Characteristics; TB, total bilirubin; TEAE, treatment-emergent adverse event; TIA, transient ischemic attack; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; UK, United Kingdom.
Job code: UK/BIL/09/25/0004|Date of preparation: September 2025